Main Window¶
When ReciPro launches, the main window appears. From this window you select the crystal, control its rotation, and invoke various functions.
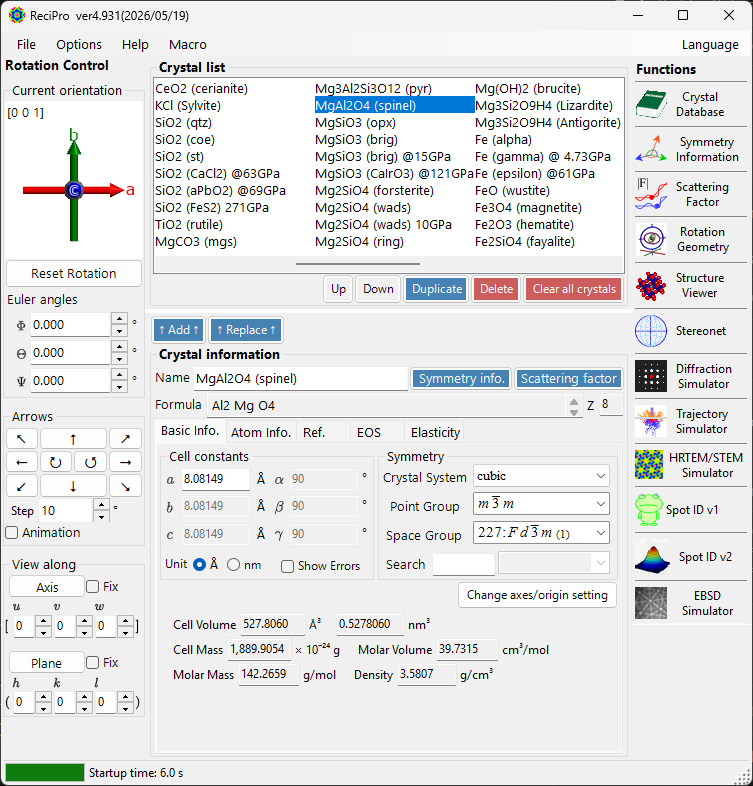
| Area | Location | Description |
|---|---|---|
| File menu | Top | File operations, options, help |
| Rotation control | Left | View/set crystal orientation |
| Crystal List | Upper centre | Select and manage crystals |
| Crystal Information | Lower centre | Edit lattice parameters, symmetry, atoms |
| Functions | Right | Launch simulation/analysis windows |
Keyboard & mouse shortcuts¶
The main window installs several application-wide shortcuts. They keep working while the Structure Viewer, Stereonet, Diffraction Simulator, Spot ID, and Calculator windows are focused.
| Shortcut | Action |
|---|---|
| F1 | Open this page of the online manual |
| CTRL+SHIFT+D | Open / close the Diffraction Simulator |
| CTRL+SHIFT+V | Open / close the Structure Viewer |
| CTRL+SHIFT+S | Open / close the Stereonet |
| CTRL+SHIFT+T | Open / close Spot ID |
| CTRL+SHIFT + arrow keys | Rotate the crystal one step in that direction (hold two arrows for a diagonal) |
| Double-tap CTRL | Open / close the Calculator |
| CTRL+SHIFT+R | Toggle the Reserved flag of the selected crystal |
| Hold CTRL while ReciPro starts | Start with OpenGL disabled (recovery for graphics problems) |
| Left-drag the orientation widget (bottom-left, under Current Direction) | Rotate the crystal |
| Right double-click the orientation widget | Copy the widget image to the clipboard |
| Single-click a function button | Open / close that window |
| Double-click a function button | Force the window visible and bring it to the front |
| Right-click a crystal in the list | Context menu (Rename / Duplicate / Delete / Export CIF…) |
| Double-click the Current Index label | Show / hide the max-UVW box |
| Drop a file on the window | Load a crystal list (.xml, .cdb2) or a crystal (.cif, .amc) |
→ See 21. Keyboard & mouse shortcuts for every window at a glance.
Basic Workflow¶
If you are new to ReciPro, refer to the following steps:
- Select the target crystal in Crystal List. To use a CIF/AMC file, drag and drop it into Crystal Information.
- If you edit lattice parameters or atom positions, press Add or Replace so the changes are written back to the crystal list.
- Set the crystal orientation in Rotation control using a zone axis, crystal plane, Euler angles, or mouse dragging.
- Open the desired tool from Functions. Diffraction, HRTEM/STEM, EBSD, and other calculation windows use the currently selected crystal and orientation.
File menu¶
File¶
| Menu item | Description |
|---|---|
| Read crystal list (as new list) | Load a crystal list file (*.xml), replacing the current list |
| Read crystal list (and add) | Append to the current list |
| Read initial crystal list | Reload the default crystal list |
| Save crystal list | Save the current crystal list |
| Export selected crystal to CIF | Save in CIF format |
| Clear crystal list | Remove all crystals |
| Exit | Close the application |
Option¶
| Menu item | Description |
|---|---|
| Show Tooltips | Toggle tooltip display |
| Use Miller-Bravais (hkil) index | Use 4-index notation for trigonal/hexagonal systems throughout the app |
| Reset registry settings on exit (effective after restart) | Reset settings on next restart |
| Disable Crystallography.Native library (requires restart) | Fall back to managed code if the native (C++) library fails to load |
| Disable all OpenGL rendering (requires restart) | For older GPUs / remote desktop |
| Disable OpenGL text rendering (requires restart) | Workaround for text-rendering issues on some GPUs |
| Use MKL Library | Use Intel MKL for numerical routines |
| Dark mode | Switch between light and dark colour themes |
| Powder diffraction function (under development) | Enable the polycrystalline (powder) diffraction window |
| Capture GUI Components… | Developer tool for saving GUI screenshots |
Help¶
| Menu item | Description |
|---|---|
| Program updates | Check whether a new version of ReciPro is available and install it |
| Hint | Display usage hints (deprecated) |
| Version history | Open the version-history dialog |
| License | Display the MIT licence |
| GitHub page | Open the ReciPro repository in a browser |
| Report bugs, requests, or comments | Open the GitHub Issues page |
| Help (Web) | Open the online manual on GitHub Pages, in the page matching the UI language. |
Language is switched from the separate Language menu (English/Japanese, requires restart).
Language¶
Switch the UI language between English and Japanese. The change takes effect after restarting ReciPro.
Macro¶
Open the Macro window to automate ReciPro operations with Python-style scripts. For repeated workflows, see the built-in functions and macro examples.
Crystal orientation control¶
The crystal's rotation state is shared by the diffraction simulator, Structure Viewer, Stereonet, HRTEM/STEM simulator, EBSD simulator, and other windows. It is not just a view setting — it defines the incident beam direction and the crystal coordinate relationship used by the simulations. A short video tutorial is available on the How to use page.
Current orientation¶
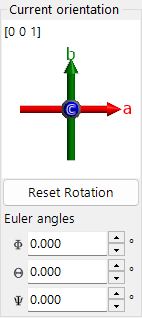
Shows crystal orientation. Drag to rotate. Axes: red = a, green = b, blue = c.
Reset rotation¶
Resets to initial: c-axis perpendicular to screen, b-axis upward.
Zone axis¶
Displays closest zone axis to screen normal (e.g., u+v+w < 30).
Euler angles (Z-X-Z)¶
Set the crystal orientation with Z–X–Z Euler angles:
- \(\Phi\): Z-axis rotation
- \(\Theta\): X-axis rotation
- \(\Psi\): Z-axis rotation
The rotations are applied in the order \(\Psi \to \Theta \to \Phi\). See Rotation Geometry and Appendix A1. Coordinate System for details.
Arrows¶
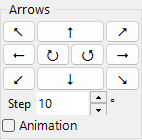
Rotate by angle Step. Check Animation for continuous rotation.
View along¶
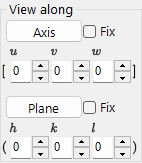
Align a zone axis [uvw] or crystal plane (hkl) perpendicular to the screen.
- Fix: when checked, the specified zone axis or plane is held spatially fixed during subsequent rotation operations.
- Axis: places the entered zone axis \([uvw]\) perpendicular to the screen. If Plane is also set, that direction is pointed upward on the screen.
- Plane: places the normal of the entered crystal plane \((hkl)\) perpendicular to the screen. If Axis is also set, that direction is pointed upward on the screen.
Basic ways to set the orientation¶
| Method | Use when | Where |
|---|---|---|
| Mouse drag | You want to rotate freely while watching the crystal axes. | Current orientation panel |
| Arrow buttons | You want small, repeatable rotations. | Arrows panel |
| Zone axis | You know the viewing direction, such as \([001]\) or \([110]\). | View along / zone-axis input |
| Plane normal | You want a crystal plane \((hkl)\) normal to the screen. | View along / plane input |
| Euler angles | You need a reproducible numeric orientation. | Euler angles (Z-X-Z) |
See Rotation Geometry and Appendix A1. Coordinate systems for the rotation matrices and coordinate conventions.
Crystal List¶
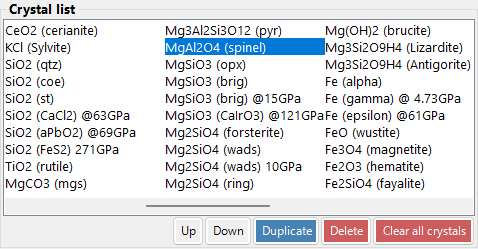
~80 crystals in default installation. Select to view details and set for calculations. Right-click a crystal in the Crystal List for a context menu: Rename, Export as CIF, Duplicate, Delete.

| Button | Action |
|---|---|
| Up / Down | Reorder |
| Duplicate | Copy the selected crystal |
| Delete / All clear | Remove crystals |
| Add / Replace | Add to list or replace the selected entry |
Crystal Information¶
Edit lattice parameters, symmetry, and atoms; drag & drop CIF/AMC files to load a structure. This control is shared by ReciPro, PDIndexer, and CSmanager, but the tabs and features shown differ per application. ReciPro shows the Basic Info, Atom, and Reference tabs (the EOS, Elasticity, and other tabs are for the other applications and are not shown in ReciPro).
Important: Press Add or Replace to save changes.
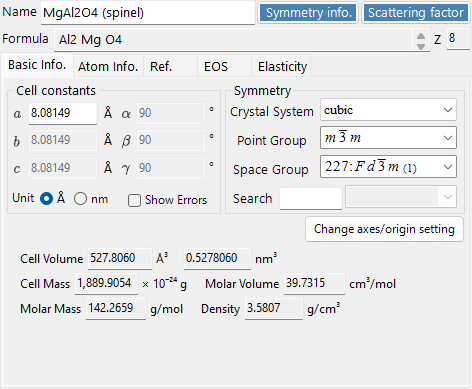
The top of the panel always shows Name (crystal name), Formula (chemical formula, computed from the atom list), and Reset (clear all fields).
Basic Info tab¶
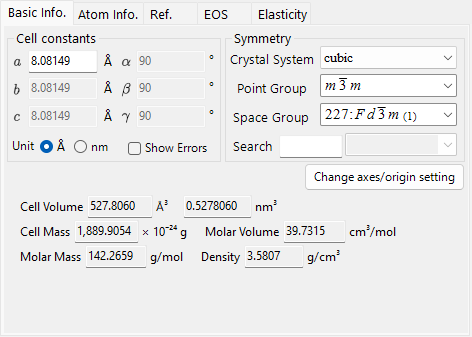
Lattice parameters, symmetry, and quantities derived from them.
| Item | Description |
|---|---|
| Cell constants | Lattice parameters a, b, c (in Å = 10⁻¹⁰ m) and α, β, γ. Choosing a symmetry constrains them automatically (e.g. a=b=c, α=β=γ=90° for cubic). |
| Symmetry | Choose the crystal system, point group, and space group. Type in the Search box to list matching candidates (case-sensitive). |
| Cell Volume / Cell Mass | Volume and mass of the unit cell. |
| Molar Volume / Molar Mass / Z / Density | Molar volume, molar mass, number of formula units per unit cell (Z), and density. Shown only when atoms have been entered. |
| Color of Profile | Color used when plotting this crystal's diffraction profile. |
Atom tab¶
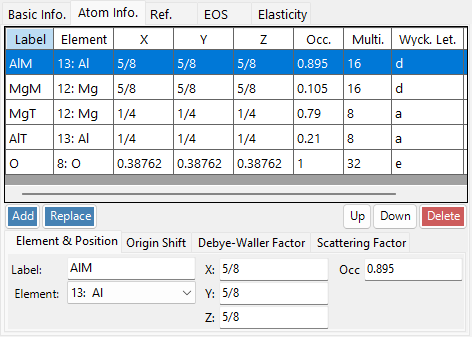
Set the species, position, temperature factor, and scattering factor of each atom. Edit the atom list with Add, Replace (replace the selected row), Up/Down (reorder), and Delete. Each atom has:
| Item | Description |
|---|---|
| Label | Atom label (any identifier). |
| Element | Element (including ionic valence). |
| X, Y, Z | Fractional coordinates (0–1). Fractions such as 1/2 or 2/3 may be entered. |
| Occ | Occupancy (0–1). |
Right-click an atom row and choose Show equivalent atom positions to open a small window listing all positions generated by the space-group symmetry operations; its Copy button copies the list as tab-separated text (e.g. for pasting into a spreadsheet).
Origin shift: shifts the origin of all atomic coordinates. Use the preset buttons (+ / −) for standard shifts, or Apply custom shift for an arbitrary amount.
Debye–Waller factor (temperature factor):
| Item | Description |
|---|---|
| Notation | Use the U or B notation. |
| Model | Isotropic or anisotropic. |
| B##, U## | For the anisotropic case, enter each component (B11, …). |
Scattering factor: choose the scattering factor used for each atom.
| Radiation | Source / setting |
|---|---|
| X-ray | Scattering factors including ionic valence (International Tables for Crystallography, Vol. C). |
| Electron | Electron scattering factors (Peng 1998, Acta Cryst. A54, 481–485). |
| Neutron | Neutron scattering lengths. Choose Natural isotope abundance or Custom isotope abundance (an arbitrary isotope composition). |
Reference tab¶
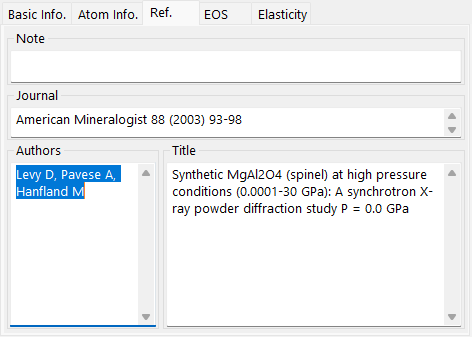
Record the source of the structure: Note, Authors, Journal, and Title.
Context menu (right-click)¶
Right-click an empty area of the control for these main actions:
| Menu item | Action |
|---|---|
| Beam Interaction | Open the Beam Interaction window. |
| Symmetry information | Open the Symmetry Information window. |
| Import from CIF, AMC | Load a crystal from a CIF / AMC file. |
| Export to CIF | Export the current crystal as CIF. |
| Revert cell constants | Restore the cell constants to the values first loaded. |
| Convert to P1 spacegroup | Expand the structure to space group P1. |
| Convert to a superstructure | Convert to a superstructure with integer multiples of a, b, c (size dialog). |
| Convert to an equivalent space group | Convert to an equivalent space group (a different axis setting). |
Functions panel¶
The vertical button strip on the right launches the analysis and simulation windows (see the Functions table below).
| Button | Description | Details |
|---|---|---|
| Crystal Database | Search and import crystals from the bundled / online databases | 1. Crystal database |
| Symmetry Information | Space-group info and ITC Vol. A symmetry diagrams | 2. Symmetry information |
| Beam Interaction | Beam–crystal interaction: reflections, attenuation, scattering factors, fluorescence | 3. Beam interaction |
| Rotation Geometry | 3D rotation matrix / goniometer angles | 4. Rotation Geometry |
| Structure Viewer | 3D crystal structure | 5. Structure viewer |
| Stereonet | Stereographic projection | 6. Stereonet |
| Diffraction Simulator | Single-crystal X-ray / electron diffraction | 7. Diffraction simulator |
| Trajectory Simulator | Monte-Carlo electron-trajectory simulation | 8. Electron trajectory |
| HRTEM/STEM Simulator | HRTEM / STEM image simulation | 9. HRTEM/STEM simulator |
| Spot ID v1 | SAED pattern indexing (formerly "TEM ID") | 10. Spot ID v1 |
| Spot ID v2 | Spot detection & indexing | 11. Spot ID v2 |
| EBSD Simulator | EBSD pattern simulation | 12. EBSD simulation |
| Powder Diffraction | Polycrystalline (powder) diffraction — enable via Option ▸ Powder diffraction function | - |